Aging Brain and Neurodegeneration
Laboratory of Molecular Physiology
Cell and Molecular Biology Program
Department of Medicine and Life Sciences
Edifici PRBB (campus del Mar)
Doctor Aiguader, 88
08003 Barcelona
(+34) 93 316 08 54
The group led by Dr. F.J. Muñoz studies the physiological and pathophysiological regulation of amyloid precursor protein (APP) cleavage by nitric oxide and oxidative stress mainly in glutamatergic hippocampal neurons. They have demonstrated that amyloid toxicity is directly dependent on nitro-oxidative stress in vascular and neuronal cells, establishing a link between extracellular amyloid deposits and intracellular neurofibrillary tangles due to the nitrotyrosination of the triose phosphate isomerase. They also found that oxidative stress triggers in both neurons and vascular brain myocytes the transcription and translation of BACE1 (the key enzyme in Aß production) by JNK and p38 MAPK pathways. They have shown that cellular stress increases BACE1 expression by the activation of PKR kinase. Furthermore, they point to a physiological role of BACE1 in the initiation and maintenance of LTP in the hippocampus mediated by the action of the kinase HRI.

BACE1 is a key enzyme for amyloid ß-peptide (Aß) production, a crucial event in Alzheimer’s disease (AD) pathogenesis, but its physiological function is practically unknown. Different cellular stressors activate BACE1 translation through the phosphorylation of the eukaryotic initiation factor-2a (eIF2a) by GCN2, PERK or PKR kinases leading to amyloidogenesis. We investigate the role of the eIF2a kinase HRI, an enzyme activated by nitric oxide (NO). NO activates HRI phosphorylation of eIF2a and induces BACE1 expression in hippocampal neurons, which allows synaptic spine growth. Consequently HRI inhibition in mice resulted in memory impairment.

Oxidative stress, a process increased with aging, activates the transcription of BACE1 through the intracellular signaling pathways JNK and MAPK p38. The increased expression of BACE1 is controlled at the transductional levels by sensor activated kinases such as PKR that phosphorylates eIF2a. Therefore PKR activation produces uncontrolled BACE1 expression and high amounts of both Aß40 and Aß42, findings that would be contributing to Alzheimer disease onset.
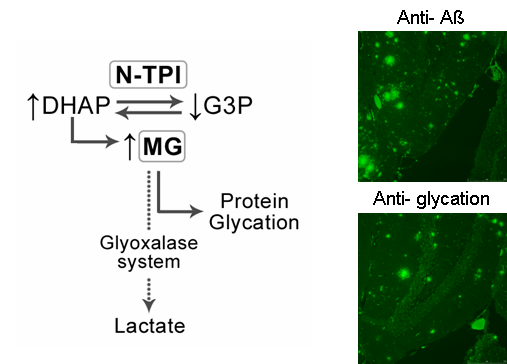
The production of free radicals generated by fibrillar aggregates of Aß leads to massive oxidative stress. Aß also activates NO production which reacts with superoxide anion to form peroxynitrite that nitrotyrosine proteins, mainly the triose phosphate isomerase (TPI). It produces the toxic methylglyoxal (MG). We have demonstrated that MG is an intracellular effector of neuronal apoptosis in AD through the activation of caspase-3 and bax, and the decrease of bcl-2 and mitochondrial.