Ion Channels
José Manuel Fernández Fernández
Group website
Research Outline
Our research focuses on the structure-function relationship of ion channels and the study of their regulation by drugs, neurotransmitters, sex hormones, intracellular proteins, glycosylation, and changes in the osmotic and physical properties of the external environment, with special emphasis on the identification of the underlying signaling pathways. During the last two years (2019-2020) our work aimed to translate our understanding of ion channels molecular physiology into a better understanding of the mechanisms of human disorders (channelopathies) affecting either the nervous system (rare hereditary forms of migraine, related paroxystic processes such as stroke-like episodes, and cerebellar dysfunctions) or the vascular system.
Current Projects
- Development of a yeast model for the study of congenital defects of glycosylation of proteins located in the Endoplasmic Reticulum and Golgi.
Aim: validation of a yeast model for those proteins involved in congenital disorders of glycosylation (CDG) that are conserved through evolution. The work will be specifically focused on mutations in the gene RFT1.
- Hypoglycosylation of CaV2.1 and Piezo channels: new pathological mechanisms and therapeutic targets for neurological disorders associated with phosphomannomutase 2 deficiency.
Aim: Understanding the pathophysiology of phosphomannomutase deficiency (PMM2-CDG), by analyzing the role of hypoglycosylation of ion channels in the development of stroke-like episodes and cerebellar syndrome associated with this rare pediatric disease.
- Low-throughput evaluation of novel, selective CaV2.1 inhibitors in animal models of Familial Hemiplegic Migraine (FHM): modulation of excitatory neurotransmission and analysis of potential therapeutic value.
Aim: Search for new pharmacological tools to treat FHM and related neurological disorders.
Team during 2019-20
-
PhD students: Albert Edo Pérez.
-
Postdocs: Mercè Izquierdo Serra.
-
Technicians: Cristina Plata Fernández.
Selected publications
- Martínez-Monseny AF, Casas-Alba D, Izquierdo-Serra M, Edo A, Bolasell M, Conejo D, Martorell L, Nascimento A, Ortez CI, Fernández-Fernández JM*, Serrano M* (Submitted) CACNA1A mutations causing early onset ataxia may associate specific facial gestalt. (*These authors have contributed equally to this work and both should be considered senior researchers, directors of the research to whom correspondence should be directed).
- Martín P, Moncada M, Castillo K, Orsi F, Ducca G, Fernández-Fernández JM, Gonzalez C, Milesi V (2021 January 6) Arachidonic acid effect on the allosteric gating mechanism of BK (Slo1) channels associated with the β1 subunit. Biochim Biophys Acta Biomembr;183550. doi: 10.1016/j.bbamem.2021.183550. PMID: 33417967. https://www.sciencedirect.com/science/article/abs/pii/S0005273621000018?via%3Dihub.
- Casas-Alba D, López-Sala L, Pérez-Ordóñez M, Mari-Vico R, Bolasell M, Martínez-Monseny AF, Muchart J, Fernández-Fernández JM, Martorell L, Serrano M (2020-2021) Early-onset severe spinocerebellar ataxia 42 with neurodevelopmental deficits (SCA42ND): Case report, pharmacological trial, and literature review. Am J Med Genet A;185(1):256-260. doi: 10.1002/ajmg.a.61939. Epub 2020 Oct 24. PMID: 33098379. https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.61939.
- Walsh S, Izquierdo-Serra M, Acosta S, Edo A, Lloret M, Moret R, Bosch E, Oliva B, Bertranpetit J*, Fernández-Fernández JM* (2020) Adaptive selection drives TRPP3 loss-of-function in an Ethiopian population. Sci Rep;10(1):20999. doi: 10.1038/s41598-020-78081-z. PMID: 33268808. https://www.nature.com/articles/s41598-020-78081-z
(*These authors have contributed equally to this work and both should be considered senior researchers, directors of the research to whom correspondence should be directed)
- Izquierdo-Serra M, Fernández-Fernández JM*, Serrano M* (2020) Rare CACNA1A mutations leading to congenital ataxia. Pflügers Arch;472(7):791-809. doi: 10.1007/s00424-020-02396-z. PMID: 32458086. https://link.springer.com/article/10.1007/s00424-020-02396-z
(*These authors have contributed equally to this work and both should be considered senior researchers, directors of the research to whom correspondence should be directed).
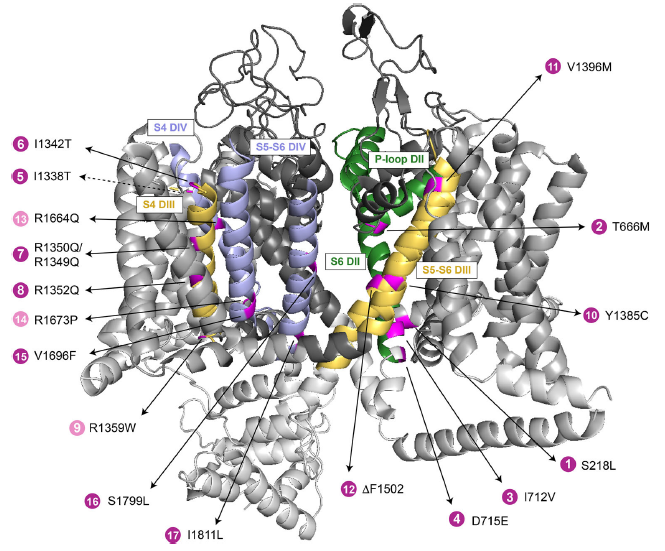
Three-dimensional location of congenital ataxia (CA)-linked CaV2.1 mutations on a CaV channel model. The structure of the α1S subunit of the CaV1.1 channel (PDB 5GJV) was used as a model considering their high level of homology. This pore-forming subunit consists of four homologous domains (DI-DIV). N- and C-termini and the intracellular loops are shown in light grey, voltage-sensor modules (S1–S4) are in grey, and Ca2+-selective pore modules (S5-P loop-S6) are in dark grey. Regions, where CA-linked mutations have been described, are colored corresponding to their domain: DII (in green, P-loop and S6), DIII (in orange, S4 and S5-P loop-S6), and DIV (in blue, S4 and S5-P loop-S6). The position of the mutation I1338T (dashed-line arrow) is approximative, as it is located in a not resolved region of the channel structure.